Bioethics
How CRISPR-Enabled Gene Editing Could Change Our World: A Huntington's Disease Case Study
Because Huntington’s disease is caused by a single gene mutation, it is a prime early target for clinical trials involving somatic cell genome editing.

{kind=link}
Inside the O’Briens is a work of fiction by Lisa Genova that chronicles several years in the life of Joe O’Brien and his family. At the age of forty-four, Joe, a Boston police officer, is diagnosed with Huntington’s disease, a progressive brain disorder. Symptoms of Huntington’s disease usually appear in a person’s thirties or forties and initially include involuntary jerking and twitching movements, as well as subtle emotional difficulties and disorganized thinking. As the disease progresses, problems develop with motor skills like walking, coordination, and balance; the involuntary jerking and twitching become more pronounced; and cognitive abilities and emotions become further impaired. While there are medications to help alleviate these symptoms, there is no cure. Most people with Huntington’s disease die of this illness fifteen to twenty years after the onset of symptoms.
The diagnosis of Huntington’s disease is only the beginning of Joe’s nightmare. At the same time that Joe learns he has this fatal disorder, he is made to understand that each of his four adult children—JJ, Patrick, Meghan and Katie—has a 50 percent chance of having inherited the disease from him.

Joe’s physician asks him if he would like to participate in a clinical trial. As Genova writes: “Joe doesn’t know the first thing about science.” He does know, however, that “He’d chop off his own head and donate it to science right now if it’d save his kids. ‘I’ll do it. Whatever it is. Sign me up.’” Later in the story, Joe’s son JJ, who has since tested gene-positive for Huntington’s disease (meaning that he will develop the disease and may pass it on to his children) shares a poignant moment with his father. Choked with emotion following the birth of his firstborn son, he tells his dad, “I’d lie down in traffic for him right now.”
JJ is not the only one of Joe’s children who is gene-positive. Joe’s daughter Meghan has been tested, and learned that she too will eventually get Huntington’s disease and experience serious physical, psychiatric and cognitive challenges, as well as an early death. Joe’s wife, Rosie, breaks down in tears: “I think about their funeral services, their beautiful faces and their bodies in caskets, buried in the ground, and I don’t want to spend one minute on this earth knowing two of my children are buried beneath it.”
Imagine, just for a moment, that you are the parent of a child with a diagnosis of a fatal disease. What would you do to save your child’s life? When a child has a fatal illness, it is common for distraught parents to say they would offer up their lives in exchange for their child’s life. Knowing this kind of bartering is only wishful thinking, however, they may also pray for a miracle. Joe’s prayers start with this plea: God, please help the scientists find a cure for HD [Huntington’s disease] so my children don’t lose their lives to this. God, please let Patrick and Katie and baby Joseph be gene negative. God, please let JJ and Meghan be cured, and let me live long enough to know they’ll be okay. Or, if there can’t be a cure yet, let them not become symptomatic until they’re much older.
While some parents pray, other parents dream. They dream of an alternate reality in which their child’s death is not inevitable. For Jane Mervar, the wife and mother of patients diagnosed with Huntington’s disease, that dream has a name: CRISPR. Unlike Joe O’Brien, Jane Mervar is not a fictional character.
In 2002, Jane Mervar’s six-year-old daughter, Karli Mukka, was diagnosed with juvenile Huntington’s disease, a rare variation that afflicts people younger than twenty. Six weeks after Karli’s diagnosis, Jane’s husband, Karl Mukka, thirty-five years old, was diagnosed with Huntington’s disease. Two years later, in 2004, Karli’s sister Jacey was diagnosed with juvenile Huntington’s disease. She was thirteen years old at the time. Not long after that, in 2007, her sister Erica was diagnosed, at age seventeen. In 2010, eight years after the onset of symptoms, Karli and Karl died within weeks of each other, leaving Jane to care for her daughters Jacey and Erica. “CRISPR is my dream,” says Jane.
 Quillette
Quillette
* * *
The human genome is widely referred to as the “book of life.” For this reason, the familiar concept of editing has been used as a metaphor to describe the work of “finding and replacing” parts of the genetic code in terms of “cutting and pasting” some of the letters or words in the book of life. Using CRISPR technology, scientists can remove, add or alter the DNA (the genome) of living organisms. So it is that Jennifer Doudna, one of the co-discoverers of the CRISPR genome-editing technology, predicts a future in which the genome is as “malleable as a piece of literary prose at the mercy of the editor’s red pen.”
How does it work? In 1993 the Spanish microbiologist Francisco Mojica discovered (and later, in 2001, named) CRISPR. In 2012, the American biochemist Jennifer Doudna and the French microbiologist and geneticist Emmanuelle Charpentier discovered a way of using CRISPR to alter the DNA of living cells. Other researchers working independently but at the same time—including Feng Zhang, a Chinese-American biochemist at the Broad Institute of MIT and Harvard and George Church of Harvard Medical School—showed that CRISPR genome editing could work with mammalian cells.
When combined with the Cas9 enzyme, the CRISPR/Cas9 genome editing system is routinely described as faster, more accurate, more efficient, and cheaper than previous genome-editing technologies—specifically, zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs).
To understand what CRISPR may offer by way of a cure for patients and prevention of disease transmission, some background about human genetics will be helpful. Deoxyribonucleic acid (DNA) is a long molecule in the shape of a double helix that encodes biological information—for example, instructions for making proteins. DNA is made up of four chemical bases—adenine (A), cytosine (C), guanine (G) and thymine (T)—which are arranged in twos to form the millions of base pairs in a DNA molecule. Adenine always pairs with thymine, and cytosine always pairs with guanine. These chemical letters spell out an organism’s genetic code.
A gene is a section of DNA on a chromosome that controls the expression of various traits such as eye color, cognitive abilities and disease risk. Humans have two copies of each gene; one copy is inherited from each biological parent. In this way, genes are the means by which genetic information is passed down through the generations from parents to offspring. The gene copies inherited from each parent can be the same or different. For example, a person may inherit two genes for brown eyes, or a gene for brown eyes and a gene for hazel eyes. Different copies of a gene are known as alleles. Sometimes differences between genes are not just about variation (for example, eye color), but can be the result of a mutation resulting in genetic disease. When this is the case, the proteins the genes normally code for may have limited or no function.
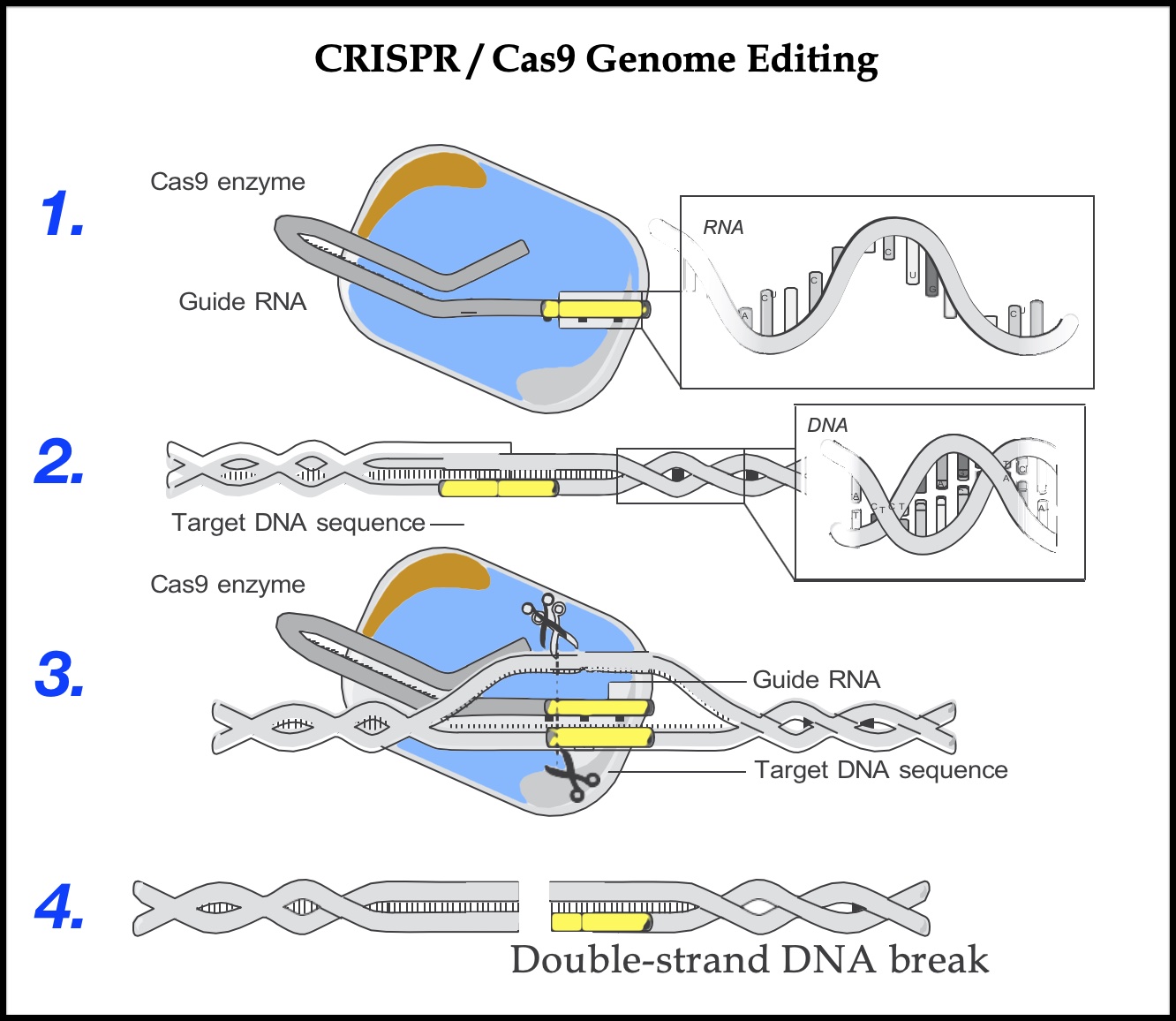
The CRISPR/Cas9 genome-editing system consists of two key molecules: a single-strand RNA called a guide RNA (known as the CRISPR), and an enzyme called Cas9 (often described as molecular scissors). RNA, like DNA, is made up of four chemical bases. Instead of thymine (T), however, RNA has a base called uracil (U), and whereas DNA is a double strand, RNA is a single strand. The guide RNA includes a short sequence of about twenty bases that is designed to complement a target DNA sequence. The RNA can pair like Velcro with one strand of the double-strand DNA. With genome editing, the single-strand guide RNA “guides” the Cas9 “molecular scissors” to the precise point in the genome where the double-strand DNA will be cut. When the guide RNA finds the target DNA sequence, it inserts itself between the two strands of the double helix and pulls them apart; the Cas9 enzyme then cuts across both strands of DNA. This break triggers the cell’s DNA repair machinery that scientists can then harness to make changes to the target DNA sequence.
* * *
Huntington’s disease is a single-gene neurodegenerative disorder (a genetic disease caused by abnormalities in a single gene). The frequency of this disease varies around the world, with the highest frequencies found in Europe and countries of European origin such as the United States and Australia. The World Health Organization estimates that in Western countries there are between fifty and seventy cases per million people.
Keep reading
On the Brink Again

They See Dead People

The Oldest Tradition in Art

The Tobacco Wars

Omer Bartov: What Went Wrong?

The Patriarchal Mind Virus
