Medicine
Treating Huntington’s
A new gene therapy that relies on gain-of-function research promises a breakthrough in neurodegenerative medicine and provides new hope for sufferers from a horrific disease.

{kind=link}
During my neurology rotation as a medical student, one of my first patients was in the middle stage of much-dreaded Huntington’s disease (HD), a hereditary, relentlessly progressive brain disorder that strips away movement, memory, and personality. It has been described as a cruel fusion of Alzheimer’s, Parkinson’s, and motor neuron diseases. HD is hereditary, and affected families have often felt hopeless, but last month, a group of English researchers, in collaboration with biotech company uniQure reported that an experimental gene therapy has dramatically slowed the course of the illness.
The Genetic and Molecular Basis of HD
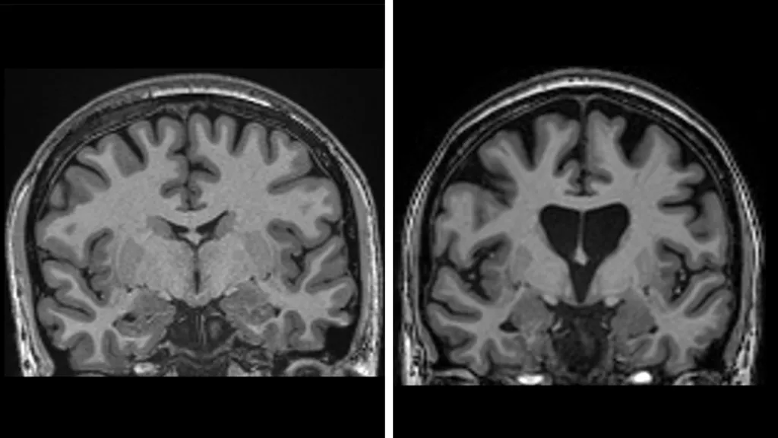
HD is inherited as autosomal dominant, which means that each child of an affected parent has a fifty percent chance of inheriting the mutation. The mutation, which is caused by a DNA repeat expansion in the HTT gene on chromosome 4, is an extended sequence of nucleotides (the building blocks of DNA) called CAG (cytosine-adenine-guanine). That abnormal DNA sequence expresses a toxic form of the huntingtin protein that accumulates and gradually damages and kills neurons in parts of the brain, especially in the caudate and putamen parts of the basal ganglia.
The Symptoms of HD
The symptoms most often begin in patients between ages thirty and sixty, while juvenile HD, which begins at earlier than twenty years of age, is rarer and tends to progress faster. Average survival is about 15–20 years after the onset of motor symptoms. The downhill course is characteristically grim.
The symptoms are of three general types:
- Motor: chorea (involuntary, jerky movements), clumsiness, dystonia, impaired balance and eye movements; later, rigidity and swallowing difficulties.
- Cognitive: slowed thinking, impaired executive function, difficulty with planning and multitasking, eventual dementia.
- Psychiatric/behavioural: depression, irritability, anxiety, apathy; sometimes obsessive–compulsive features or psychosis. Sleep disruption and weight loss are common.
The diagnosis is made on the basis of a clinical examination plus genetic testing for the expanded HTT CAG repeat, which provides definitive evidence of the condition. For those at risk but without symptoms, predictive genetic testing is available, and is usually accompanied by extensive counselling.
The UK research team became emotional as they described the results of their successful clinical trial last month: The gene therapy treatment slowed the progression of the disease by 75 percent, which means that the degree of decline that would normally be expected in one year would take four years to appear after treatment. That affords patients decades of “good quality life,” Professor Sarah Tabrizi told BBC News.
The new treatment, a type of gene therapy administered during lengthy and delicate brain surgery, is a tour de force of neurosurgery and molecular biology. The latter aspect involves cutting edge gene therapy used to interdict the functioning of an aberrant gene.
How, exactly, does the therapy work? In short, its goal is to reduce levels of the toxic huntingtin protein permanently, in a single treatment. It starts with a safe, genetically modified virus, AMT-130, that has been altered to contain a specially designed sequence of DNA that is the active agent. But delivering it to the right place is difficult: It requires a surgical operation during which the virus is infused deep into the brain using real-time MRI scanning to guide a microcatheter to two brain regions—the caudate nucleus and the putamen. This typically takes 12–18 hours of neurosurgery.
The virus then acts like a molecular messenger that delivers the new piece of DNA into brain cells, where the biological magic occurs: The virus carries a specially designed piece of DNA that instructs the neurons to produce RNA that prevents synthesis of the mutant protein. More specifically, the newly-transformed cells begin to produce a small fragment of genetic material (microRNA) that is designed to intercept and disable the instructions (in messenger RNA) being sent from the patient’s brain cells’ DNA to synthesise mutant huntingtin protein. (Recall how proteins are synthesised: In a cell’s nucleus, DNA is transcribed into messenger RNA (mRNA), which is then transcribed into protein in the cellular cytoplasm.)
The virus is removed by the brain’s immune system in a matter of days, but the DNA is expected to remain in the patients’ neurons for the rest of their lives. This would permanently lower levels of the mutant huntingtin protein in the brain—and therein lies the magic.
The Clinical Trial
Results from the trial—which involved 26 US patients—were released in a statement by uniQure, the sponsor, which said, in part:
The study met its prespecified primary endpoint, with high-dose AMT-130 demonstrating a statistically significant slowing of disease progression as measured by the composite Unified Huntington’s Disease Rating Scale (cUHDRS) at 36 months compared to a propensity score-matched external control. The study also met a key secondary endpoint by achieving statistically significant slowing of disease progression as measured by Total Functional Capacity (TFC) at 36 months compared to a propensity score-matched external control.
The pivotal finding was that three years after surgery there was an average 75 percent slowing of the disease, based on a formula that combines cognition, motor function, and the ability to manage the activities of daily living.
The results also show that the treatment preserves brain cells. Levels of neurofilaments in spinal fluid—a clear sign of brain cell death—would be expected to increase by about a third if the disease had continued to progress, but they were actually lower than at the start of the trial. This is an important complement to the finding of slowed clinical manifestations of disease progression.
“This is the result we’ve been waiting for,” said Professor Ed Wild, consultant neurologist at the National Hospital for Neurology and Neurosurgery in London, which is part of the UCLH NHS Foundation Trust. “There was every chance that we would never see a result like this, so to be living in a world where we know this is not only possible, but the actual magnitude of the effect is breathtaking, it’s very difficult to fully encapsulate the emotion.”
The treatment was considered safe, although some patients did develop inflammation from the virus that caused headaches and confusion that either resolved or needed steroid treatment. According to the statement from uniQure:
AMT-130 was generally well-tolerated, with a manageable safety profile at both doses. As of June 30, 2025 [when data collection for the current report stopped], no new drug-related serious adverse events have been observed since December 2022. The most common adverse events in the treatment groups were related to the administration procedure, which all resolved.
Approximately 75,000 people have Huntington’s disease in the US and around a further 7,000 in the UK, and 2,160 in Australia. Hundreds of thousands of additional people carry the mutation and are therefore expected to develop the disease.
Keep reading
Dark Artist

The Passion of Bess McNeill


Capturing the ‘Odyssey’
Biting the Hand